The COVID-19 pandemic has accelerated consumer, clinician, and regulator acceptance of digital health products across the healthcare ecosystem – including the use of 366+ unique digital endpoints in new medical product development (representing a 929% increase in the use of digital endpoints from 2019-2022), AI/ML-enabled products and modern-day software-based technologies for the use in a variety of therapeutic areas in clinical research and patient care. Such development in the field is exciting, but these advances bring to the surface several questions, such as,
- “How do providers and consumers differentiate between the 300k+ health apps available for download and more sophisticated, evidence-based digital health solutions?”;
- “What are the evidentiary requirements for regulated digital health products? How do we define these standards to meet the needs of a broad group of healthcare decision-makers and end-users?”;
- “How can we ensure inclusivity is table stakes in the development of digital health solutions so that we are capturing insights from a diverse group of patients?”; and
- “How do we best design studies to evaluate the safety and efficacy of digital health products in a way that ensures generalizability and effective translation in real-world care settings.”
To address these questions, the U.S. Food and Drug Administration (FDA) has not only taken a leadership role across global regulatory agencies but is also making substantial progress nationally. FDA is heading into October after releasing several landmark program and guidance updates that show progress in implementing the agency’s Digital Health Innovation Action Plan and promise for the field of digital health as a whole. These updates came amidst a push in Washington to reauthorize PDUFA legislation, which contains several provisions that impact digital health, including a plan to produce a cross-FDA framework to guide the generation and use of digitally-derived data for regulatory decision-making. It is a pivotal time to get things right from the beginning, especially in the developing field of digital health, and DiMe is setting a precedent in lockstep with our regulatory partners.
Setting a Precedent: Regulatory Oversight and the Software-Product Flood
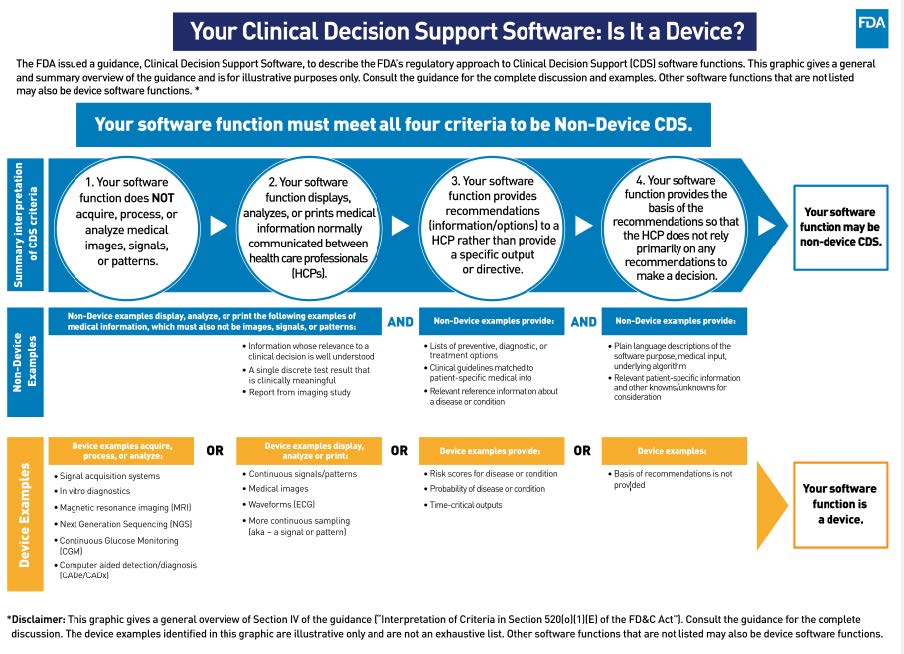
Last month, the FDA released the long-anticipated final version of its guidance on the use of Clinical Decision Support (CDS) Software, a key piece in its overall framework for regulating digital health technologies and software as a medical device (SaMD). The guidance is intended to clarify what types of software are subject to FDA regulation. It contains a set of criteria against which the industry can assess the regulatory status of its products. It also showcases examples of AI/ML products that FDA should regulate, including devices to predict sepsis, identify patient deterioration, forecast heart failure hospitalizations, flag patients with a risk of opioid addiction, and more. The CDS guidance will generate ripple effects across the tech and health sectors. This includes large-scale changes in how artificial intelligence and software embedded in electronic health record systems, which previously did not need clearance from FDA, are regulated and used to support safe and effective care delivery.
Findings from a Five-Year Pilot to Alternate Regulatory Approach: What’s Next?
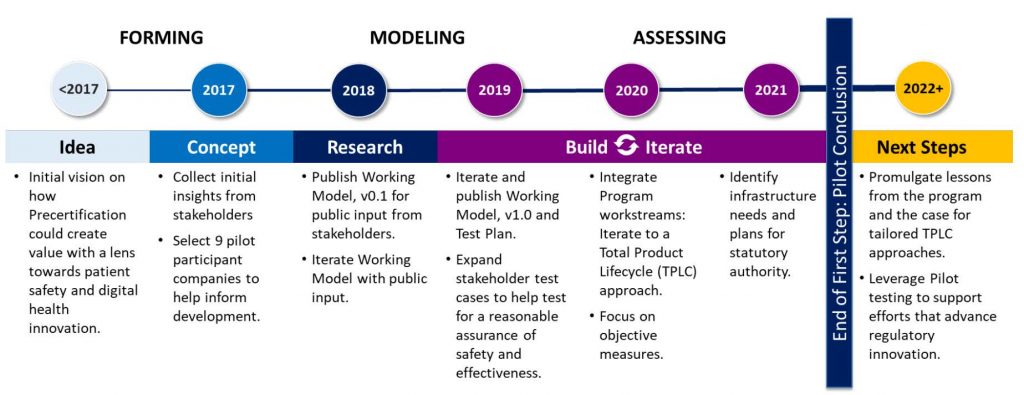
FDA also released the results of the completion of its Digital Health Software Precertification (Pre-Cert) Pilot Program in late September. The pilot program (2017-2022) was designed to evaluate an adaptive, alternate approach to the regulatory oversight of medical device software. It was centered on the pre-certification of digital health companies that met specific criteria, as opposed to the clearance of singular digital health tools submitted to the agency by those companies for review. In its report, FDA found that the piloted approach to regulatory oversight helped keep pace with rapidly evolving digital health technologies and continuous software updates. Still, continuing the program beyond the pilot phase would require legislative action. Congress should act quickly on the heels of pilot program completion to expand FDA’s ability to regulate the safe and effective use of digital health products to keep pace with tech timelines and meet end-user needs.
Navigating a New Policy Environment: What’s Inside or Outside FDA’s Oversight?
To help product developers, many of which have not previously marketed an FDA-regulated product, apply the new guidance and understand program updates, FDA has released a Digital Health Policy navigator tool. The interactive tool outlines whether the software part of a digital product is subject to FDA oversight alongside a suite of relevant laws, guidelines, and policies to consider as a part of product development. This is a clear signal of the Agency’s willingness to build bridges with the innovator community and to clarify its approach to the regulatory oversight of new and existing technology intended for medical purposes. The development of this tool also promotes a standards-driven approach to innovation, ensuring patient safety and aiding innovators in developing products that consistently improve patients’ lives.
DiMe is Building the Future of Digital Healthcare with FDA
Digital health solutions offer enormous promise to address some of the most pressing and persistent challenges in healthcare. DiMe, alongside industry experts, examines the progress, gaps, and building tools and resources with a new collaborative, “Digital Health Regulatory Pathways,” launched in June 2022. The multi-sector collaboration convenes organizations and experts from Abbott, Abbvie, Aetion, American Medical Association (AMA), Amgen, AstraZeneca, Consumer Technology Association (CTA), Digital Therapeutic Alliance (DTA), U.S. Food and Drug Administration (FDA), Genentech, a member of the Roche Group, Google, Harvard-MIT Center for Regulatory Science, Janssen, Otsuka Pharmaceuticals, Rock Health, Sidekick Health, and Tidepool. The collaboration will build resources and tools to support innovators seeking to optimize their regulatory strategy to drive the development of high-quality, trustworthy digital health products that best meet their commercial goals and the needs of patients.
DiMe and FDA’s collaborative work also expands to a variety of areas to accelerate the ethical, effective, equitable, and safe use of digital medicine that include:
- Measuring Digital Health: DiMe hosts DATAcc, an FDA collaborative community, to maximize digital health measures to improve health outcomes, health economics, and health equity by deploying toolkits and frameworks for the development, design, and deployment of digital health products.
- Driving inclusion: FDA is currently participating in DiMe’s Diversity, Equity, and Inclusion in Digitized Clinical Trials project to develop best practices, practical resources, and educational materials to design clinical trials that successfully engage, enroll, and retain underrepresented populations.
- Building the foundation for remote monitoring: DiMe’s new resource The Playbook: Digital Healthcare Edition builds upon The Playbook: Digital Clinical Measures, which was developed with 30 industry partners, including FDA and the European Medicines Agency, to accelerate research, enhance clinical care, and improve public health.
- Integrating sensor data: DiMe recently launched resources to advance Sensor Data Integrations, with the participation from FDA, to help realize the promise of sensor-generated data to drive better decisions faster and to improve healthcare delivery and research.
- Serving as an expert: With deep expertise in digital health innovation, DiMe serves as part of FDA’s Network of Experts Program to advise the Agency and engage closely on various digital health and digital medicine developments.
To fully realize the benefits of advances in novel digital offerings, concomitant innovation in regulatory science is essential. With the DiMe and FDA’s partnership, we are committed to clarifying regulatory approaches for emerging digital health technologies and facilitating optimized regulatory oversight for high-quality digital health products through collaborative programming. This programming, along with FDA’s diligent implementation of its digital health action plan, is a giant step towards standards setting for digital medicine and, ultimately, towards optimizing human health through the digital transformation of research and care delivery.


